2020-02-17 10:38


扫码打开虎嗅APP

文章来自微信公众号:返朴(ID:fanpu2019),作者:Chuan Xiao等,翻译:徐颖、顾舒晨
2020年1月31日,印度理工学院德里分校的研究人员在bioRxiv上发表了一篇论文,称新型冠状病毒是人为改造的病毒,因其刺突蛋白S蛋白上含有四个插入片段,这些片段与HIV-1的某些片段完全同源或具有相似性。
这篇未经审稿的论文掀起了诸多讨论,虽然已被撤稿,但依旧给人们留下了疑问。2月14日,杜克大学医学中心的Chuan Xiao等人在Emerging Microbes & Infections杂志上发文,分析了新冠病毒中这四个插入片段与hiv-1等其他基因的同源性,驳斥了2019-nCoV可能是通过人为获得HIV-1基因组中的一些基因片段而产生的“生化武器”这种观点。

当一种新的病原体肆虐人类世界之时,探寻它的起源往往是一个亟待解决的关键问题。了解病原体的来源对阻止其进一步传播以及加快疫苗研发等都是至关重要的,特别是对那些跨越宿主障碍的动物源性传染病来说。例如目前已为人熟知艾滋病病毒(HIV-1) [1]、非典型性肺炎病毒(SARS)[2]以及中东呼吸综合征冠状病毒(MERS)[3]等均是典型的例子。
然而,揭示一种新的人类病原体的来源需要广泛而有力的科学证据,这是一个非常复杂的过程,往往耗时经年,不幸的是,在确认新病原体的真正来源之前,总有些阴谋论抢先浮出水面,称新病原体是人为制造的。然而历史终将给出答案,阴谋论终将被科学推翻。
2019年12月,中国首次报道了一种新型致病性冠状病毒,这一病毒迅速扩散到25个国家。目前,它已经感染了45000多人,并已造成了1000多人死亡(http://2019ncov.Chinacdc.Cn/2019-Ncov)。科学家们仅用了两周左右的时间就完成了这种新病毒基因组的测序工作,并于1月12日公布了结果[4]。
1月13日,世界卫生组织 (WHO) 将这种新的病毒暂命名为2019-nCoV。进化分析显示2019-nCoV是冠状病毒的新成员,能够感染人类。基因分析表明2019-nCoV与冠状病毒同源,但它不同于引起非典(SARS)和中东呼吸综合征(MERS)的冠状病毒[5, 6],而是与2013年从云南蝙蝠身上分离到的蝙蝠冠状病毒RaTG13具有高度的遗传相似性 (96.3%) ,这表明类RaTG13病毒很可能是当前2019-nCoV的源头,但并非直接来源[7]。
由于暂时无法获知2019-nCoV的确切来源,人们开始猜测2019-nCoV可能来源于人为的基因改造,甚至有人认为这可能是一种生物武器。对此,媒体纷纷出来辟谣。然而,最近的一项发表在预印本上的非正式的研究报告表明,与其他冠状病毒相比,在2019-nCoV的刺突糖蛋白中有四个插入片段,而刺突糖蛋白正是病毒进入靶细胞的关键蛋白[8]。
该研究声称,这些插入片段与来自三个不同国家 (泰国、肯尼亚和印度) 的某些独特HIV-1毒株的囊膜糖蛋白或Gag蛋白中高度可变 (V)区 (V1、V4和V5) 的基因序列相同或相似。结合蛋白结构模型分析,作者推测这些与HIV-1蛋白相似的插入片段可以增强病毒对宿主细胞上特异性受体的亲和力,并扩大2019-nCoV宿主细胞的范围。这项研究暗示2019-nCoV病毒可能是通过从HIV-1基因组获得特定基因片段而人为产生的。
我们将2019-nCoV病毒、其他冠状病毒和HIV-1病毒的序列与GenBank数据库的序列进行了仔细的比对,并没有发现这四个插入片段是HIV-1所特有的,亦或是2019-nCoV病毒从HIV-1获得了这些插入片段的证据。
首先,在GenBank数据库中对这些病毒基因进行序列比对的结果表明,前100个相同或高度同源的序列均来自哺乳动物的宿主基因、昆虫、细菌等 (表1) 。比对结果只有几个显示与冠状病毒同源,但它们均与HIV-1病毒无关。病毒序列数据库的比对结果还显示,这些序列广泛地存在于从噬菌体、流感到巨型真核病毒等不同类型的病毒中 (表1) 。
这些结果清楚地表明,插入的基因序列广泛存在于包括病毒在内的各种生物体中,绝不是HIV-1所特有的。当用2019-nCoV病毒的序列在整个数据库中进行检索时,我们发现它与冠状病毒的匹配程度更高,仅有少数与HIV-1相匹配(表1)。并且,虽然有19条记录显示插入片段1和2与HIV-1能够完全匹配,但插入片段3和4与HIV-1的匹配率很低(仅有42%到88%匹配)。
另外,插入片段4能够与HIV-1基因组中的多个不同基因片段(包括gag、pol和env)进行模糊匹配,这表明它们之间的同源性太低(低于42%),因此并不可信。此外,在HIV数据库中检索这四个插入片段,我们也能得到相似的结论(https://www.hiv.lanl.gov/components/sequence/HIV/search/search.html)。
在任何HIV-1中均未发现与插入片段3和4完全匹配的序列,这也说明插入的基因序列广泛的存在于包括病毒在内的各种生物体中,并非HIV-1所特有的。另一方面,HIV-1囊膜蛋白中与之匹配的的区域都具有高度可变性,常常存在大量的插入和缺失突变,这也表明这些片段对于HIV-1囊膜糖蛋白的生物学功能来说并不是必需的。
同时,与插入片段1和2完全匹配的序列仅能在少数的HIV-1病毒株中检测到,说明在数以万计的天然HIV-1中同时存在这四个插入突变的序列也是非常罕见甚至并不存在。这也解释了为什么这四个插入片段独立存在于不同的HIV-1基因组中的原因[8]。由于这些插入片段与HIV-1的同源性低,在HIV-1序列中也非常少见,因此我们认为HIV-1并非是2019-nCoV基因组中插入序列的来源。
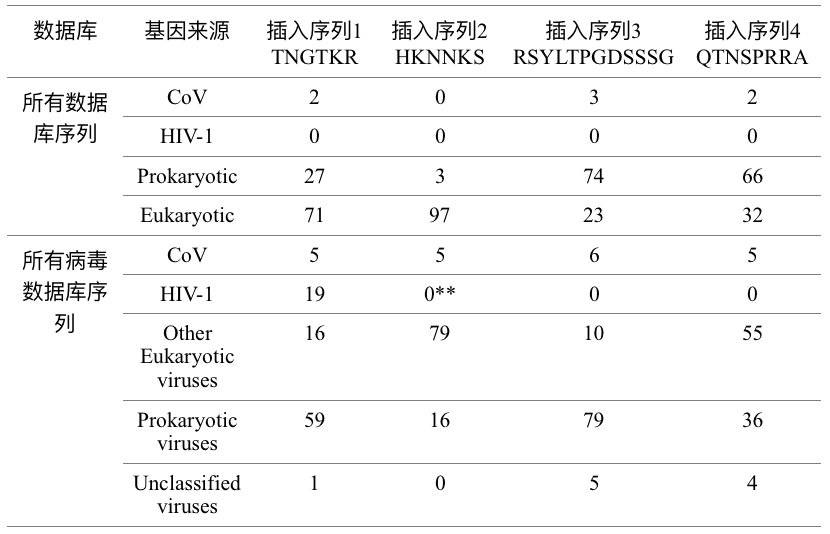
表1. 4个插入序列与序列数据库对比搜索结果
其次,这些插入片段不仅存在于2019-nCoV病毒中,还存在于其他三个源于蝙蝠的β属冠状病毒中:包括分离自浙江并于2018年上传到GenBank数据库中的ZC45和ZXC21病毒,以及于2013年分离自云南的RaTG13病毒[7]。
与ZC45和ZXC21相比,RaTG13与2019-nCoV更为相似(如图1A所示),它们的刺突蛋白的相似性为97.7%。在RaTG13基因组中,其中两个插入片段(HKNNKS和RSYLTPGDSSSSG)与2019-nCoV中的插入片段完全相同,第三个插入片段仅有一个丝氨酸(Threonine,T)到异亮氨酸(Isoleucine,I)的替换(TNGIKR),第四个插入片段的C端缺失4个氨基酸(QTNS----)(如图1B所示)。
与RaTG13相比,ZC45和ZXC21与2019-nCoV的差异更大,但是这两种病毒也有与插入片段1、2和3类似的序列(图 1B)。此外,许多其它的冠状病毒在插入片段1的位点上都有类似的插入仅序列有些许差异。这些结果清楚地表明,在2019-nCoV被发现之前,这四种插入序列中的三种均天然存在于三种蝙蝠的冠状病毒中。这无疑驳斥了2019-nCoV可能是通过人为获得HIV-1基因组中的一些基因片段而产生的“生化武器”这种观点。相反,2019-nCoV更可能来源于类RaTG13的冠状病毒。
第三,2019-nCoV中的插入片段1和2与某些HIV-1 gp120分离株中的V4和V5区域具有相同的6个氨基酸基序,它们在结构上相互接近,由LE loop将二者分隔开(如图1C所示)[9]。然而,位于2019-nCoV插入片段1和2之间的插入片段3的序列与HIV-1 gp120的V1区域相似(存在氨基酸的缺失)。V1区域位于gp120蛋白序列的另一侧,离V4和V5区域较远,因此它不可能与gp120(图1C)中的V4/V5区域具有相互作用。但是在2019-nCoV刺突蛋白的结构模型中,V1区域恰恰位于V4和V5之间[10]。
插入片段4被发现与HIV-1的Gag蛋白上区域相同,但这一蛋白与病毒的入侵无关。另外,这个插入位点与其它几个位点相距太远,无法与2019-nCoV刺突蛋白中的其他三个插入片段形成同一个的结构单元(如图1C所示)。因此,综上所述,虽然预印本的作者指出2019-nCoV 从HIV-1中获得一些结构上看似不相干的部分进而产生了一个独特的蛋白结构,该结构有利于增强刺突蛋白与受体的结合能力,但是我们并不认为这种结构有任何选择优势或理论根据[8]。
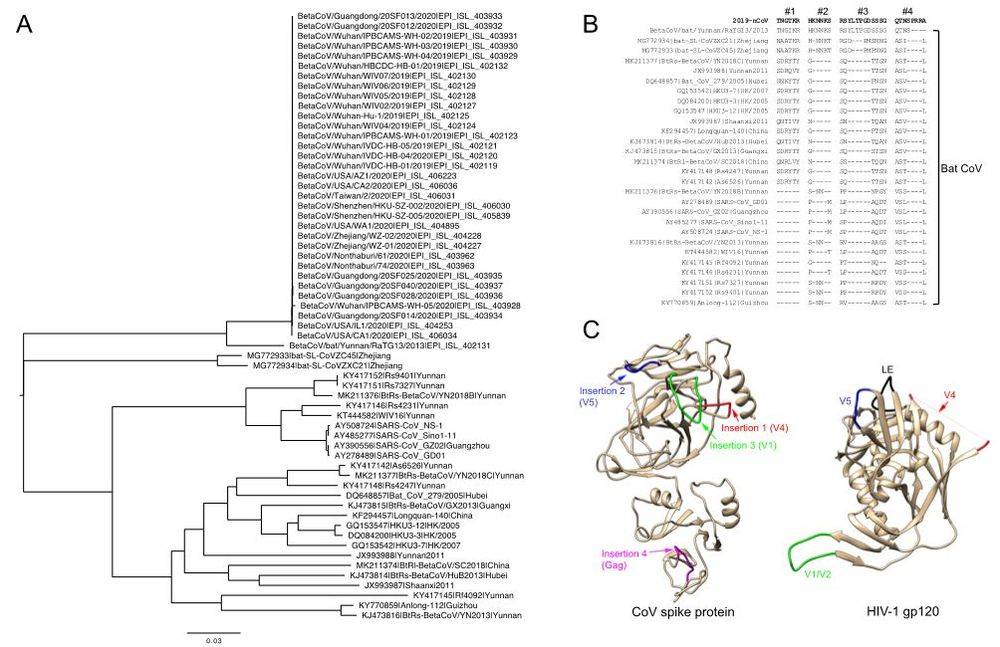
图1. 2019-nCoV和蝙蝠冠状病毒的序列和结构分析。(A)刺突基因序列的系统进化树。(B)2019-nCoV和蝙蝠冠状病毒序列之间疑似插入位点的序列比对。比对结果中氨基酸的缺失用“-”表示。插入片段的序号在比对结果的上方标示。(C)CoV刺突蛋白和HIV-1 gp120中四个插入片段的结构对比。2019-nCoV的结构使用默认参数在TASSER服务器进行建模。1~708位残基中仅有相关结构域用丝带图展示,不包括305~603位残基。这四个插入片段分别用红色、蓝色、绿色和品红色显示。HIV-1 gp120结构(PDB 1GC1)用丝带图展示。V4、V5、V1/V2和LE loop分别用红色、蓝色、绿色和黑色展示。
这三个蝙蝠冠状病毒株如何获得这些插入片段目前尚不得而知。对于病毒而言,若要从其他生物体获得额外的插入序列,通常需要它与其他生物体有直接的相互作用,通过同源或非同源重组的方式获得插入序列[11]。因此只有当两种病毒共同感染同一个细胞时,蝙蝠冠状病毒才可能从HIV-1获得基因片段。
由于蝙蝠冠状病毒和HIV-1的宿主细胞不同,因此两者交换遗传物质的机会几乎可以忽略不计。但由于这些基序广泛存在于各种哺乳动物细胞中,因此蝙蝠冠状病毒更有可能从它们感染细胞的基因组中获得这些基序并进行重组。想要回答这一问题,科研人员应该对野生动物和家畜中的冠状病毒进行更广泛的研究。
鉴定这三种蝙蝠冠状病毒和最近流行的2019-nCoV毒株中的插入片段的来源对我们理解冠状病毒如何突破宿主障碍,由感染动物跨越至感染人类、适应人类这些过程至关重要。目前的数据表明,RaTG13与2019-nCoV的关系最为密切[7]。但由于它们之间的遗传差异太大,因此RaTG13并不能作为2019-nCoV的直系祖先。其他与2019-nCoV关系更为密切的病毒,例如以果子狸为中间宿主的SARS和以骆驼为中间宿主的MERS[3, 12],是否为其直接来源仍有待研究。
人们需要更多的研究以确定2019-nCoV的真正来源。但病毒的溯源需要对各种野生动物和家畜进行筛选,这一过程可能需要很长时间。因此无论如何,减少或避免与野生动物的直接接触,对防控未来可能产生的新型流行性感染病依然至关重要。
得益于生物信息学分析工具的快速发展,现在大家都能够对新序列进行广泛快速的分析。我们必须进行全面透彻的分析才能充分理解新的基因组信息隐含的真正生物学意义。那些有偏见的、片面的以及不正确的分析所得的结论则会助长阴谋论,阻挡科学发现的进程,极大的影响公众卫生的防控工作。
文章来自微信公众号:返朴(ID:fanpu2019),作者:Chuan Xiao等,翻译:徐颖、顾舒晨

 05:28
05:28






 13:16
13:16
 11:24
11:24
 05:31
05:31
 11:43
11:43
 23:56
23:56
 07:29
07:29
 25:06
25:06
 20:37
20:37
 07:20
07:20




