
2023-01-26 14:47
抗病毒“韦”药:拟肽

扫码打开虎嗅APP

本文来自微信公众号:同写意(ID:tongxieyi),作者:杨翼,题图来自:视觉中国
一、抗病毒药物
抗病毒药物(Antiviral drugs)是一类用于治疗病毒感染的药物。大多数抗病毒药物针对特定病毒,而广谱抗病毒药物可有效对抗多种病毒。[1] 抗病毒药物是抗微生物药物(antimicrobials)的一个类别。
抗微生物药物的涵盖面更加广泛,除了抗病毒药物之外,还包括抗生素(antibiotics,也称为抗菌药物antibacterial drugs)、抗真菌药物(antifungal)和抗寄生虫药物(antiparasitic drugs)。[2]现在可用的大多数抗病毒药物针对 HIV、疱疹病毒、乙型和丙型肝炎病毒以及甲型和乙型流感病毒。[3] 以Covid-19为代表的冠状病毒,也成为了如今抗病毒药物开发的重点领域。

抗微生物药物分类,制图:写同意
二、多肽和拟肽类抗病毒药物
不难发现,很多抗病毒药物的通用名都是以vir(韦)结尾的,如果进一步观察这些“韦”系抗病毒药物的化学结构,就会发现它们中间有不少是拟肽(peptidomimetics)的形式存在的(更不消说那些以tide结尾的抗病毒药物,例如Bulevirtide),例如抗HIV病毒的Atazanavir(阿扎那韦,商品名Reyataz等)、Lopinavir(洛匹那韦,与ritonavir的FDC联合制剂Kaletra)和Nelfinavir奈非那韦(商品名Viracept);抗HCV病毒的Boceprevir(波普瑞韦,商品名Victrelis)和Telaprevir(特拉匹韦,商品名Incivek,Incivo);以及抗SARS-CoV-2病毒的联合疗法制剂nirmatrelvir/ritonavir(奈玛特韦/利托那韦,商品名Paxlovid)等(图1)。
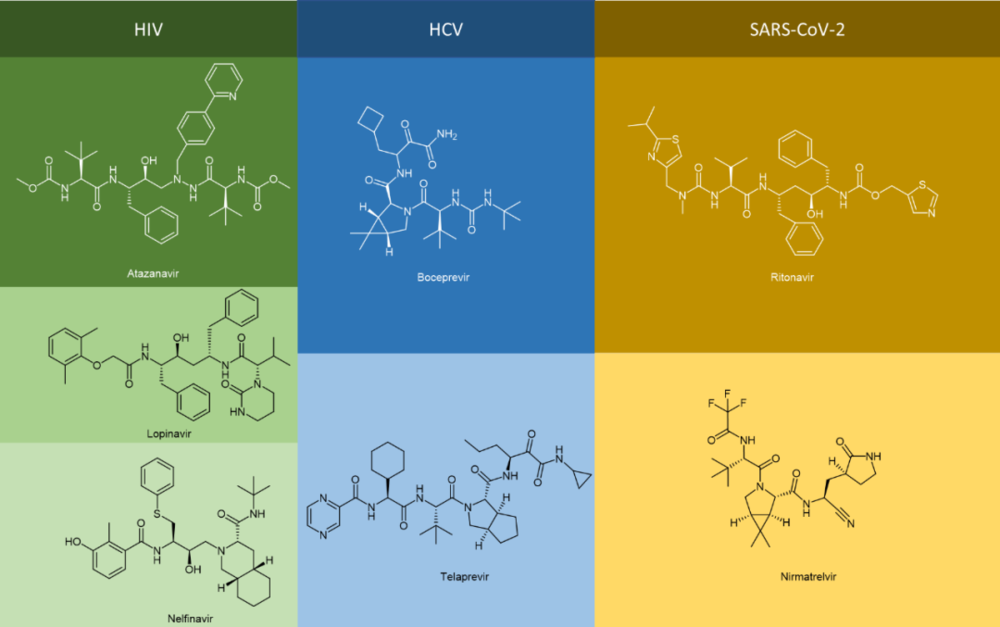
图1. 常见的拟肽类抗病毒药物分子结构
使用拟肽作为治疗药物具有很多优势,例如增强的结合力、改善的代谢稳定性和良好的生物利用度(相对于多肽类药物)。将传统的多肽类药物,通过化学改性工具库里的不同策略, 转化为拟肽类物质, 进一步提高受体亲和力并改善药代学特性,是该类别抗病毒药物的重要开发手段。
三、常见的多肽化学改性策略
传统的多肽类药物,掣肘于较差的代谢稳定性和极低的口服生物利用度,因此研发人员通过化学改性的方法,以期在以上两个领域做出显著的改善。药物设计研究人员常用的多肽化学改性手段包括以下方法:
1. 末端封闭
将多肽的N-端用乙酰基封闭,将C-端修饰为酰胺,是多肽化学改性的最小动作。这样的修饰可以降低因为Nα-介导的DKP降解(diketopiperazine)。[4]这种保护作用对于分子量较小的多肽来说意义尤其重大。截至 2021 年底,62个(不包括已撤消的罗穆尔肽和安巴莫司汀)已上市的短肽(由小于或等于 15 个氨基酸残基组成)中,有 37 个实体在其 N 端通过酰化、焦谷氨酸或环化的方式得以封闭,只有 9 种线性肽实际上具有自由Nα基团。末端封闭对于多肽的二级结构已会产生很重大的影响,因为这样会剥夺分子上所带的电荷。而多肽空间结构的变化,反过来又可以影响分子稳定性。[5]
除了这两种最常规的修饰之外,还可以在C-端加载α-keto amide(α-酮羰基酰胺)的结构,将酮羰基引入多肽分子中,使之摇身一变,转化为可以与受体形成共价结合的共价拟肽药物。[6]例如针对HCV病毒的丝氨酸蛋白酶的拟肽药物boceprevir 波普瑞韦和telaprevir特拉匹韦,它们的分子C-端就含有α-keto amide的结构(图2)。该修饰极大地提升了母体化合物的效力和药代动力学特性。
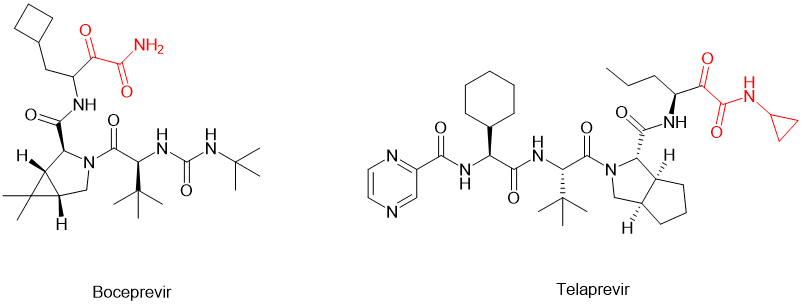
图2. 含有α-keto amide结构(红色部分)的拟肽药物boceprevir和telaprevir
另外一种日趋流行的共价药物弹头硼酸结构,也时常被引入多肽分子之中。例如在一项研究中,科学家将硼酸结构加载到了寨卡病毒、西尼罗河病毒和登革热病毒蛋白酶的二肽抑制剂中,取代原有的羧酸或酰胺基团,所衍生出的硼酸基抑制剂(图3)的亲和力相对于修饰前增加了一千倍。[7]

图3. 硼酸基寨卡病毒、西尼罗河病毒和登革热病毒蛋白酶二肽抑制剂
2. 酰胺的生物电子等排体替换(bioisostereric replacement)
多肽的骨架是由酰胺键组成的,对于多肽的物理化学性质无比重要。通过生物电子等排体(bioisostere)替换酰胺键,是研究人员“由多肽至拟肽”的一种重要策略。常见的酰胺电子等排体包括:酯类esters、硫代酰胺类 thioamides、1,2,3-三唑类1,2,3-triazoles、磺胺类sulfonamides,亚膦酰胺phosphonamidites、羟基乙烯hydroxy ethylene和羟基乙胺hydroxy ethylamine等(图4)。[8]
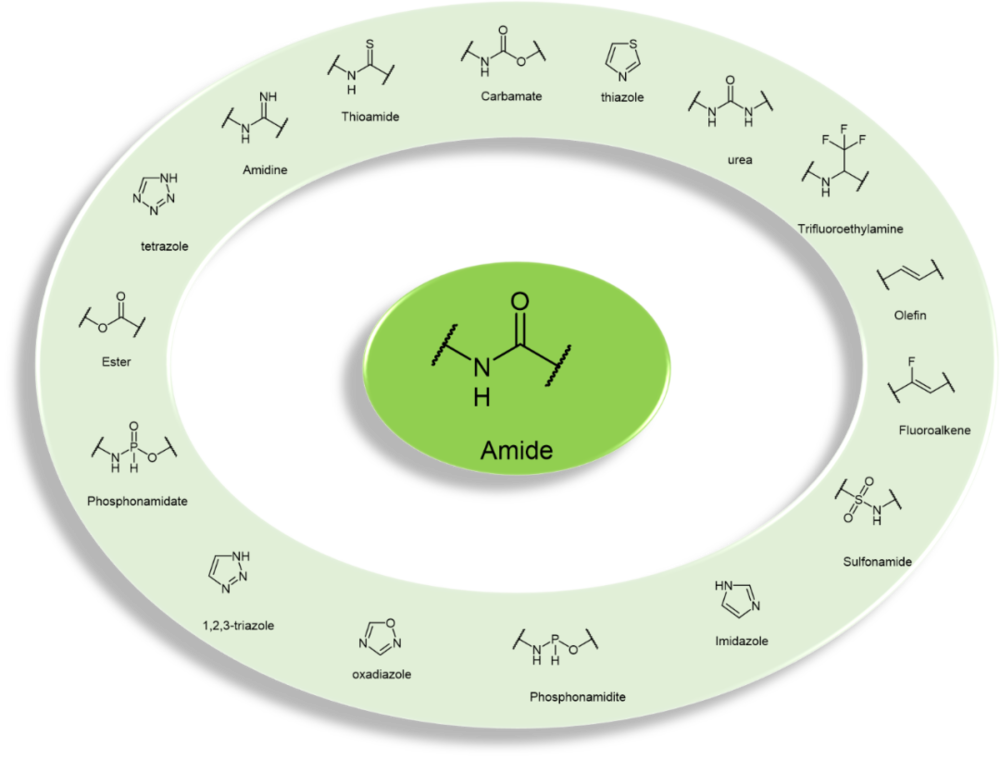
图4. 常见酰胺生物电子等排体
通过生物等排体替换酰胺键的化学修饰方法的例子,在拟肽药物开发中不胜枚举。例如第一个抗HIV病毒的蛋白酶抑制剂Saquinavir沙奎那韦,就是用羟基亚甲基的结构取代了多肽主链的不稳定的酰胺键(图5),提高了药物的代谢稳定性。[9]
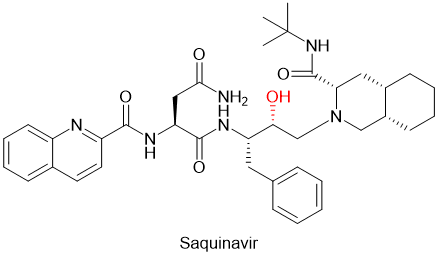
图5. Saquinavir化学结构
3. 氨基酸修饰
氨基酸修饰的方法极其广泛,包括使用non-proteinogenic氨基酸取代蛋白质氨基酸,比如用D-氨基酸取代L-氨基酸,从而提高底物的水解稳定性。研究人员拥有越来越大的非蛋白质氨基酸库,可以支持不同类型的氨基酸改性。
已上市的多肽药物中,D-氨基酸甚至成为了常规的多肽药物残基。在2021年批准的多肽药物中,就有voclosporin (D-Ala) 和odevixibat(D-4-hydroxyphenylalanine)两款含有D-氨基酸。更有甚者,2021年通过FDA审批的defelikefalin(商品名Korsuva,一种镇痛阿片肽,用于治疗中度至重度瘙痒[10])采取了全D-氨基酸(D-Phe, D-Leu, D-Lys)的策略(图6)。
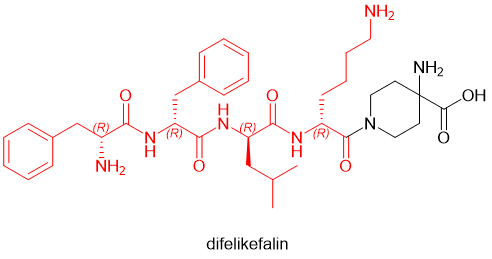
图6. 全D-型氨基酸多肽Difelikefahlin的化学结构
4. 环肽
多肽的环化从来都是重要的多肽化学改性策略,从上世纪80年代就上市的第一款多肽口服制剂cyclosporin A开始,人们就一直将多肽环化奉为提高多肽稳定性,改善药代动力学性质,提高生物利用度的重要手段之一。
截止2021年,通过FDA, EMA和日本FMDA的将近105个多肽治疗剂与诊断试剂中,环肽几乎占据了半壁江山(47%),其中单环肽的比例为37%,多环肽为10%。在抗病毒拟肽中,环肽也出现在了抗 HCV药物,特别是大环可逆药物NS3/4A 丝氨酸蛋白酶抑制剂中,例如simeprevir西美瑞韦,glecaprevir格卡瑞韦和voxilaprevir伏西瑞韦 (图7)。[11]
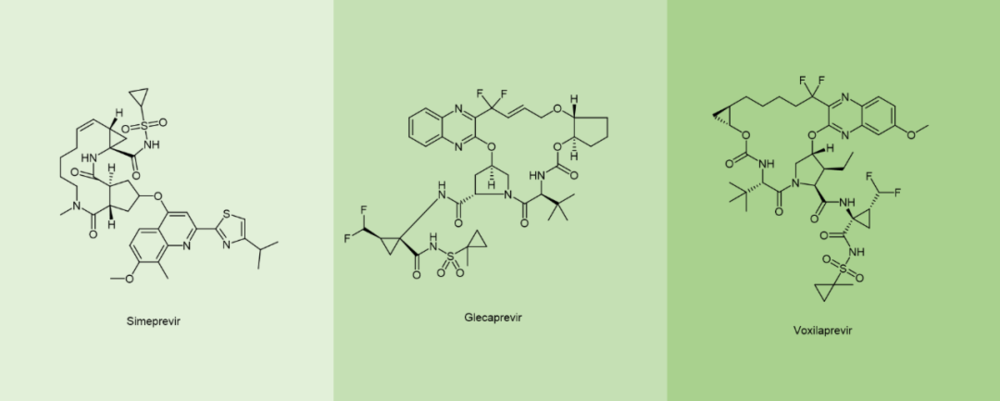
图7. 环拟肽Simeprevir, Glecaprevir, Voxilaprevir化学结构
四、抗病毒拟肽研发及产业状态
1. 抗SARS-CoV-2病毒拟肽药物
拟肽抗病毒治疗剂最受关注的例子可能莫过于Paxlovid了,它在SARS-CoV-2病毒掀起的Covid-19全球大爆发中起到了稳定局面的作用。Paxlovid是一款FDC(Fixed-dose combination, 固定剂量组合)的复方制剂,它含有两种活性物质nirmatrelvir和ritonavir,正如前文提及的那样,它们都是拟肽类物质(图1)。
其中的ritonavir早年单独作为抗HIV病毒药物上市,但因为药效和副作用的原因,很快被新生代取代。但因为它和CYP3A4酶高度的结合力,以及对CYP3A4这个单加氧化酶的抑制作用(通过不同的机理,包括(I) 代谢中间复合物 (MIC) 的形成,MIC与血红素群紧密结合;(II) 未修饰的ritonavir与血红素铁的强结合;(III) 血红素破坏;(IV) 活性ritonavir中间体与 CYP3A4 脱辅基蛋白的共价连接等[12]), ritonavir在Paxlovid中充当了药代动力学增强剂的角色。
通过它对CYP3A4的抑制作用,保护了真正的抗病毒活性物质nirmatrelvir较少受到CYP3A4的酶促降解,提高了它的半衰期和药物暴露量。Nirmatrelvir的药理学作用在于它对于3C样蛋白酶(3C-like protease, 3CLPRO,亦称Mpro)的抑制作用,nirmatrelvir因此是Mpro蛋白酶抑制剂。Mpro是一种半胱氨酸蛋白酶,它对SARS-CoV-2的刺突蛋白上的S糖蛋白的裂解作用,对于病毒与宿主细胞融合并感染细胞的生物学过程极为重要,因此抑制这个蛋白酶是Paxlovid的药理学功效的核心。
Nirmatrelvir是一款共价拟肽药物,在与受体蛋白的结合口袋通过非共价结合的基础上,nirmatrelvir上的核心构件,即氰基(cyano group)弹头(warhead, 特指共价药物上的活性亲电基团),可以与Mpro蛋白酶上的Cys145残基侧链的巯基作用,形成thioimidate(硫代亚胺酸酯),从而通过共价的方式紧密结合靶向的受体蛋白,可以在较低剂量的条件下实现高效的药效作用。
值得注意的是,Paxlovid是一款口服抗病毒药物,这正是药代动力学增强剂ritonavir,以及nirmatrelvir自身的药代学特征的基础之上实现的。

图8. 巯基化合物与腈类化合物产生硫代亚胺酸酯的反应
2. 抗HCV病毒拟肽药物
抗HCV(丙型肝炎病毒)也拟肽药物的核心任务之一。丙型肝炎是一种由丙型肝炎病毒(HCV)引起的肝脏疾病。目前,HCV感染的治疗基于直接抗病毒药物(DAA,directacting antivirals),靶向特定NS蛋白(非结构蛋白),并破坏病毒复制。
2011 年,FDA批准了第一代DAA,即 boceprevir 波普瑞韦和telaprevir特拉匹韦(图2),它们针对基因1型HCV感染患者的NS3/4A,它们的分子C-端都含有α-keto amide的结构。这些DAA与聚乙二醇干扰素(peg-IFN)和利巴韦林联合使用,成本显然不低。[13]
第二代DAAs拟肽药物daclatasvir和sofosbuvir(图9),分别作为NS5A和NS5B抑制剂得到应用,代表了HCV治疗的突破,提高了更多患者的持续病毒反应率(SVR)。[14]
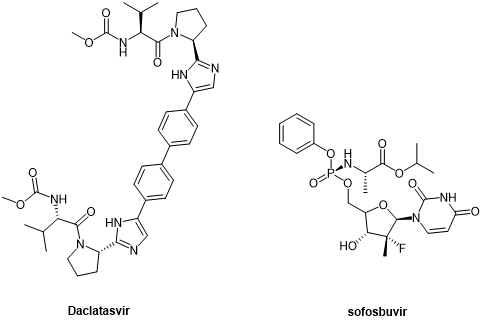
图9. 第二代抗HCV拟肽药物Daclatasvir与Sofosbuvir化学结构
Glecaprevir和Voxilaprevir(图10)是针对NS3/4A开发的第三代DAA,能够靶向所有基因型,包括今天代表HCV治疗里程碑的基因3型,它们都是环拟肽。
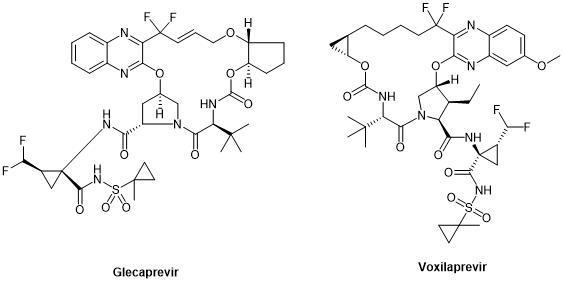
图10. 第三代抗HCV拟肽药物Glecaprevir与Voxilaprevir化学结构
3. 抗HIV病毒拟肽药物
抗HIV药物可以分为:核苷类逆转录酶抑制剂(NRTIs,Nucleoside Reverse Transcriptase Inhibitors)、非核苷类逆转录酶抑制剂(NNRTIs,Non-Nucleoside Reverse Transcriptase Inhibitors)、蛋白酶抑制剂(PI,Protease Inhibitors)、融合抑制剂(Fusion Inhibitors)、CCR5拮抗剂 (CCR5 Antagonists)、 整合酶链转移抑制剂 (INSTIs, Integrase Strand Transfer Inhibitors)、附着抑制剂(Attachment Inhibitors)和附着后抑制剂 (Post-Attachment Inhibitors)等很多类型。蛋白酶抑制剂中,很多药物属于拟肽行列。
FDA于1995年批准的拟肽沙奎那韦(saquinavir,图5)属于第一代蛋白酶抑制剂。第一代抑制剂基于羟基乙烯和羟乙胺电子等排物。[15]中心羟基通过模拟水解步骤的过渡态,结合催化性天冬氨酸残基(图11)。在Paxlovid中梅开二度的药代动力学增强剂ritonavir当年就是第一代的抗HIV蛋白酶抑制剂。
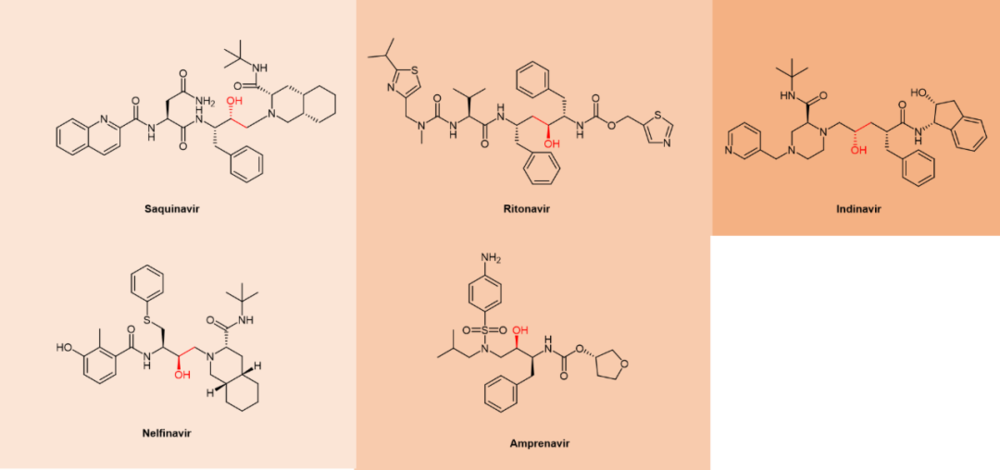
图11. 第一代抗HIV拟肽蛋白酶抑制剂
洛匹那韦(Lopinavir)由 Abbott 开发,属于第二代抗HIV蛋白酶抑制剂(图12),用于改善ritonavir的特性。洛匹那韦无法单独给药,因此成为第一个与作为药代动力学增强剂的利托那韦以联合疗法使用的复方制剂。
阿扎那韦(Atazanavir)于 2003 年获得批准,它成为第一个每天一次有效给药的蛋白酶抑制剂。其半衰期比以前的蛋白酶抑制剂更长。[16]
替拉那韦(Tipranavir)于 2005 年获得批准,并于 2008 年扩展到儿科用途。由于其结构上存在许多差异,它对某些耐药 HIV 菌株保持效力,并且似乎具有更高的遗传屏障,需要许多突变才能产生耐药性 。[17]2006年获得FDA批准的地瑞那韦(Darunavir)代表了最新的抗HIV蛋白酶抑制剂。

图12. 第二代抗HIV拟肽蛋白酶抑制剂
总结
拟肽是药物化学中的强大工具,将活性肽转化为结合了多肽与小分子药物特点的综合体,无论在药理学还是药代动力学特性方面,都较多肽药物有了显著提升,可以大规模实现口服递送的方式。在过去的十年里,人们对抗病毒拟肽的研发兴趣与日俱增,Paxlovid的出现证明了拟肽在抗病毒方面的巨大价值。
随着对病毒致病过程理解的加深,不同抗病毒药物之间的合纵连横的情况也可能成为现实,开发广谱抗病毒药物也是科学家致力的研究方向。人们有理由对于新一代“韦”抗病毒药物,它在药效、安全性、抗耐药性、递送方式等众多领域的不断优化报以信心。
参考文献:
[1] Rossignol JF Nitazoxanide: a first-in-class broad-spectrum antiviral agent. Antiviral Res. 2014, 110: 94–103.
[2] Rick Daniels; Leslie H. Nicoll. "Pharmacology – Nursing Management". Contemporary Medical-Surgical Nursing. Cengage Learning, 2011. p. 397.
[3] Kausar S, et al. A review: Mechanism of action of antiviral drugs. International Journal of Immunopathology and Pharmacology. 202135: 20587384211002621.
[4] Yang, Y. Side Reactions in Peptide Synthesis. Elsevier. 2015, 22-28.
[5] King, T.A. et al. (2021) Photocatalytic methods for amino acid modification. Chem. Soc. Rev. 50, 39–57.
[6] Robello, M. et al. The Alpha Keto Amide Moiety as a Privileged Motif in Medicinal Chemistry: Current Insights and Emerging Opportunities. J. Med. Chem. 2021, 64, 7, 3508–3545.
[7] Nitshe, C. et al. Peptide–Boronic Acid Inhibitors of Flaviviral Proteases: Medicinal Chemistry and Structural Biology. J. Med. Chem. 2017, 60, 1, 511–516.
[8] Kumari, S. et al. Amide Bond Bioisosteres: Strategies, Synthesis, and Successes. J. Med. Chem. 2020, 63, 21, 12290–12358.
[9] Ghosh, A.K. et al. Recent progress in the development of HIV-1 protease inhibitors for the treatment of HIV/AIDS. 2016, J. Med. Chem. 59, 5172–5208.
[10] Allerton, C. Pain Therapeutics: Current and Future Treatment Paradigms. Royal Society of Chemistry. 2013, pp. 56
[11] Nageswara, D. et al. (2021) Discovery of quinoxaline-based P1–P3 macrocyclic NS3/4A protease inhibitors with potent activity against drug-resistant hepatitis C virus variants. J. Med. Chem. 64, 11972–11989.
[12] Loos, N, H. et al. The Mechanism-Based Inactivation of CYP3A4 by Ritonavir: What Mechanism? Int. J. Mol. Sci. 2022, 23, 9866.
[13] Kjellin, M. et al. The effect of the first-generation HCV-protease inhibitors boceprevir and telaprevir and the relation to baseline NS3 resistance mutations in genotype 1: experience from a small Swedish cohort. Ups. J. Med. Sci. 2018, 123 (1), 50−56.
[14] Wei, L. et al. Long-term outcomes in patients with chronic hepatitis C in the current era of direct-acting antiviral agents. Expert Rev. Anti Infect Ther. 2019, 17 (5), 311−325.
[15] Ghosh, A. K. et al. The FDA Approved HIV-1 Protease Inhibitors for Treatment of HIV/AIDS. In Burger’s Medicinal Chemistry and Drug Discovery, 7th ed.; Abraham, D. J., Rotella, D. P., Eds.; John Wiley & Sons: Hoboken, NJ, 2010; Vol. 7, pp 1−74.
[16] Bold, G.; et al. New aza-dipeptide analogues as potent and orally absorbed HIV-1 protease inhibitors: candidates for clinical development. J. Med. Chem. 1998, 41, 3387−3401.
[17] Doyon, L.; et al. Selection and characterization of HIV-1 showing reduced susceptibility to the non-peptidic protease inhibitor tipranavir. Antiviral Res. 2005, 68, 27−35.
本文来自微信公众号:同写意(ID:tongxieyi),作者:杨翼

 07:02
07:02









 08:14
08:14
 22:05
22:05
 03:08
03:08
 13:16
13:16
 13:50
13:50
 04:16
04:16
 25:06
25:06
 01:24
01:24
 20:37
20:37

